Lupine Publishers| Journal of Biomedical Engineering and Biosciences

Abstract
In this study, Metronidazole (Met) and it’s modified derivatives are optimized by employing density functional theory with B3LYP/6-31g (d,p) level theory to explore their structural and thermodynamical properties. Molecular electrostatic potential (MEP) calculation has performed to calculate their possible electrophilic and nucleophilic attack. ADMET prediction was performed to search the absorption, metabolism and toxic level. Finally, this study can be helpful to design a potent candidate.
Keywords: Metronidazole; Density functional theory; HOMO-LUMO; MEP; ADMET
Abbrevations:Met: Metronidazole; DFT: Density functional theory; HOMO: Highest occupied molecular orbital; LUMO: Lowest unoccupied molecular orbital; MEP: Molecular electrostatic potential; ADMET: Absorption, distribution, metabolism, excretion, and toxicity
Introduction
Metronidazole (Met) is an antibiotic [1] and antiprotozoal drug [2]. It is widely used in the treatment of amoebiasis, trichomoniasis and giardiasis [3,4]. It has some demerits depending on the type and nature of unusual physical condition and on the limit of dose. High and long term dose can cause of leucopenia, neutropenia and peripheral neuropathy diseases [5]. Adverse effect and resistance of drugs indicate the importance of the discovery of new potential candidate. In computer aided drug design system, physicochemical, molecular docking, nonbonding interactions, and ADMET predictions are important criteria to evaluate newly designed molecules [6]. Drug modification is another alternative way to search better agent, which can increase the selective action of drug and reduce the side effect. Recently, it has been seen the trait of modifying drugs using halogens and alkyl group play important role in improving drug performance [7].
Herein, I report the optimization of Metronidazole (Met) and its modified derivatives to investigate their biochemical behavior on the basis of quantum mechanical approach. The free energy, electronic energy, enthalpy, dipole moment, HOMO-LUMO gap, hardness, softness, chemical potential and electrostatic potential have been calculated. All the newly designed derivatives show better thermodynamic properties, and some of them exhibit better chemical reactivity than parent drug. From the regarding quantum chemical studies, it’s assuming that, some of the designed compounds may have profound effect as drug.
Methods and Materials
Computational Details
In computer aided drug design, quantum mechanical methods are widely used to predict thermal, molecular orbital, and molecular electrostatic potential properties [8]. Initial geometry of Metronidazole (Met) was taken from the online structure database named ChemSpider [9]. Geometry optimization and further modification of all structures carried out using Gaussian 09 program [10]. Density functional theory (DFT) with Becke’s (B) [11] threeparameter hybrid model, Lee, Yang and Parr’s (LYP) correlation functional [12] under Pople’s 6-31g (d,p) basis set has been employed to optimize and elucidate their thermal and molecular orbital properties [13]. Initial optimization of all compounds was performed in the gas phase. Dipole moment, electronic energy, enthalpy, free energy and electrostatic potential are calculated for all the compounds (Figure 1).
Figure 1: Chemical structure of Metronidazole (Met) and its modified analogues.

Frontier molecular orbital features HOMO (highest occupied molecular orbital), LUMO (lowest unoccupied molecular orbital) were calculated at the same level of theory. For each of the drugs, HOMO-LUMO energy gap, hardness (η), softness (S) and chemical potential were calculated from the energies of frontier HOMO and LUMO as reported considering Parr and Pearson interpretation [14,15] of DFT and Koopmans theorem [16] on the correlation of ionization potential and electron affinities with HOMO and LUMO energy (𝜀). The following equations are used to calculate hardness (η), softness (S) and chemical potential (μ);
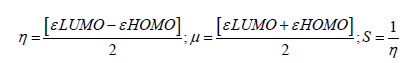
In computer aided drug discovery system, computational predictions are using to explore absorption, distribution, metabolism, excretion, and toxicity (ADMET) which saves on time and investment. AdmetSAR online database was utilized to predict ADMET properties of Metronidazole and its analogues [17].
Result and Discussion
Thermodynamic Analysis
Simple modifications of molecular structure significantly influence the structural properties including thermal and molecular orbital parameters. From the free energy, and enthalpy values, spontaneity of a reaction and stability of a product can be predicted [18]. In drug design, hydrogen bond formation and nonbonded interactions also influenced by dipole moment. Increased dipole moment can improve the binding property [19]. From thermodynamic data (Table 1), the free energy of Metronidazole is -623.7600 Hartree, where M1 shows the highest negative value (-921.4882 Hartree). The –F substitution (M1) influence the free energy significantly. Highly negative free energy is favourable for stable configuration. Again, the dipole moment of Metronidazole is 4.1174 Debye where M3 shows the maximum dipole moment (5.3559 Debye) due to substitution of –NH2 group (Figure 2).
Table 1: The stoichiometry, molecular weight, electronic energy, enthalpy, free energy in Hartree and dipole moment (Debye) of Metronidazole (Met) and its analogues.

Figure 2: Most stable optimized structures of Metronidazole (Met) and newly designed analogues. Optimized with B3LYP/6- 31g (d, p) level theory.

Molecular Orbital Properties
Table 2: Energy (eV) of HOMO, LUMO, gap, hardness, softness and chemical potential of the designed drugs.

The HOMO-LUMO energies, hardness, softness, chemical potential of all compounds is presented in Table 2. The electronic absorption relates to the transition from the ground state to the first excited state and mainly described by one electron excitation from HOMO to LUMO [20]. The chemical hardness, softness, and potential values depend on the energy gap of HOMO-LUMO [21,22]. Kinetic stability decreases with the decrease of HOMO-LUMO gap. As a result, removal of electrons from ground state HOMO to excited state LUMO requires less energy. In our studies, Metronidazole shows the HOMO-LUMO gap 4.6124 eV, where M3 have the lowest energy gap (4.1783 eV) with highest softness (0.4787 eV) which may contribute higher chemical reactivity (Figure 3).
Figure 3: Frontier molecular orbital (HOMO-LUMO) and related energy of Metronidazole (Met) and M3.

Molecular Electrostatic Potential Analysis
Molecular electrostatic potential (MEP) was calculated at B3LYP/6-31G (d,p) level of theory to forecast the reactive sites for electrophilic and nucleophilic attack of all optimized structures [23]. Red color represents maximum negative area which favorable site for electrophilic attack, blue color indicates the maximum positive area which favorable site for nucleophilic attack and green color represent zero potential area. MEP displays molecular size, shape as well as positive, negative and neutral electrostatic potential regions simultaneously in terms of color grading. It is seen from MEP map, region having the negative potential are over electronegative atom (oxygen atoms) and having positive potential are over hydrogen atoms. Here, the maximum negative potentiality is found for M3 is -0.3497 a.u (deepest red) for oxygen atoms and the highest positive potentiality of M1 is +0.3903 a.u (deepest blue) of hydrogen atoms.
Read More About Lupine Publishers Journal of Biomedical Engineering and Biosciences Please Click on Below Link: https://biomedical-sciences-lupine-publishers.blogspot.com/
No comments:
Post a Comment
Note: only a member of this blog may post a comment.